
Clinical Reasoning: Pes cavus and neuropathy
Neurology. 2019 Aug 20;93(8):e823-e826. DOI:10.1212/WNL.0000000000007976.
Pes cavusをみたらCharcot-Marie-Tooth病とまず第一に考えますが,しっかりと全体像を評価して検討する必要があると感じました.
Session 1
症例
18歳 女性.
ニューロパチーと振戦に関して神経クリニックに紹介.
神経疾患の家族歴なし.
出生~成長発達
正期産.処女歩行は14ヶ月と軽度の遅れ.
つま先歩きがぎこちなく,走る際に周りについていくのが難しかった.
7歳でADHDと診断され,土踏まずが高く,踵歩きが困難.
初期診断
筋電図と神経伝導検査は脱髄性ニューロパチーの所見があり,Charcot-Marie-Tooth病(CMT)と診断.
初期診断後の経過
その後,数年かけて症状が進行した.躓きやすく,時に転倒し,走ることが困難になった.手すりなしで階段をのぼることができたが,下るのは注意を要した.左優位の手の振戦が出現し,物に手を近づけると悪化した.感覚障害やしびれ,膀胱直腸障害,聴覚,視覚の障害はなかった.高校を卒業し大学進学した.
神経診察
知的には問題なく,脳神経麻痺も認めない.
眼球運動障害や球麻痺は認めなかった.
足関節拘縮とpes cavusを認めた.軽度の下肢痙性があるが,萎縮やfasciculationは認めず,短母指外転筋や第一背側骨間筋,前脛骨筋,長母趾伸筋などの遠位筋では5-/5の筋力低下があった.腱反射は上肢で2+,下肢では3+.足底反射は伸展.
触覚や痛覚,温度書く,位置角は正常であったが,つま先の振動覚は軽度低下していた.
左優位の企図振戦や指鼻試験での軽度の測定障害を認めた.反復拮抗運動障害は明らかではなかった.
歩行は軽度wide-basedで,継脚歩行はできなかった.
Questions
- 鑑別疾患は?
- 鑑別を進めるために有用な検査は?
Section 2
本例は下肢で上位運動ニューロン徴候があり,小脳失調や振動覚低下,pes cavusを認めた.
pes cavus(凹足)の考察
high archesによる足の変形で,ニューロパチーの長期罹患(先天性,後天性とも)で生じる.遺伝性痙性対麻痺,Friedreich失調症,脊柱管癒合不全(二分脊椎,脊髄係留症候群)などで生じる.CMTでよくみられる.
だが,通常,脱髄型CMTでは本例のような腱反射亢進はみられない.CMTでは遠位での筋力低下,筋萎縮,感覚障害,腱反射消失を呈する.錐体路徴候を呈する稀なCMTの病型もある.
本例での局在
脊髄後外側(皮質脊髄路+後索),tremor circuit(両側深部小脳核,対側の視床腹外側核へ投射する小脳遠心路),脱髄性ポリニューロパチー,ミエロニューロパチーなどを考える.
痛みや感覚レベル,膀胱直腸障害がないことから圧迫性脊髄症らしくない.
本例での鑑別疾患
若年発症であり遺伝性の非圧迫性脊髄症を考慮する.
- 遺伝性痙性対麻痺(HSP):遺伝性疾患で,皮質脊髄路のlength-dependentな軸索変性で,上位運動ニューロン徴候を呈する.HSPは,純粋型(下肢痙性や腱反射亢進,足底反射,排尿症状,遠位部での振動覚障害)と複合型(ニューロパチー,痙攣,パーキンソニズム,認知機能障害,失調,筋萎縮,低身長,視覚障害,聴覚各障害)がある.
HSPは時に,小脳失調やニューロパチー,運動ニューロン疾患,認知障害,白質脳症などの他の障害を合併する.本例では,臨床的には複合型HSPである(下肢錐体路徴候,小脳失調,ニューロパチー).
- Friedreich失調症:最もcommonな常染色体劣性遺伝形式の失調症で,GAAリピート延長が96%である.平均発症年齢が10~15歳で,失調歩行や錐体路徴候(痙性と足底反射伸展),軸索型感覚ニューロパチー(関節位置覚と振動覚障害,腱反射消失)を呈する.Friedreich失調症では腱反射が保たれることもあるが,脱髄性ニューロパチーは通常生じない.
- 副腎白質ジストロフィー:女性のX-linked副腎白質ジストロフィー保因者は脊髄ニューロパチーを呈する.
- その他:企図振戦は多発性硬化症,脊髄小脳変性症,種々の変性疾患,代謝性,腫瘍性など深部小脳核やその遠心路を傷害する病変で生じる.
本例では,上位運動ニューロン徴候と下位運動ニューロン徴候,小脳症状もみられ,遅発性白質脳症などの代謝性疾患が示唆された.白質脳症は多様な表現系や遺伝疾患を含む疾患群である.脱髄性ニューロパチーがあることから鑑別疾患を狭めることができる.
X-linked 副腎白質ジストロフィー(女性での副腎脊髄ニューロパチー),Krabbe病(KD),異染性白質ジストロフィー,脳腱黄色腫症(治療可能な疾患なので,仮に腱黄色腫がなくてもコレスタノール値を測定すべきである),Pelizaeus- Merzbacher-like disease.
検査所見
血液検査
血算,ビタミンB12,ビタミンE,乳酸,肝腎機能,甲状腺,CRP,ホモシステイン,メチルマロン酸,銅は正常.
極長鎖脂肪酸は正常(副腎脊髄ニューロパチーらしくはないが,女性保因者では正常値となることもある).
コレスタノールは未測定.
腰椎穿刺
施行していない.
画像検査
- 脊椎MRI:正常.
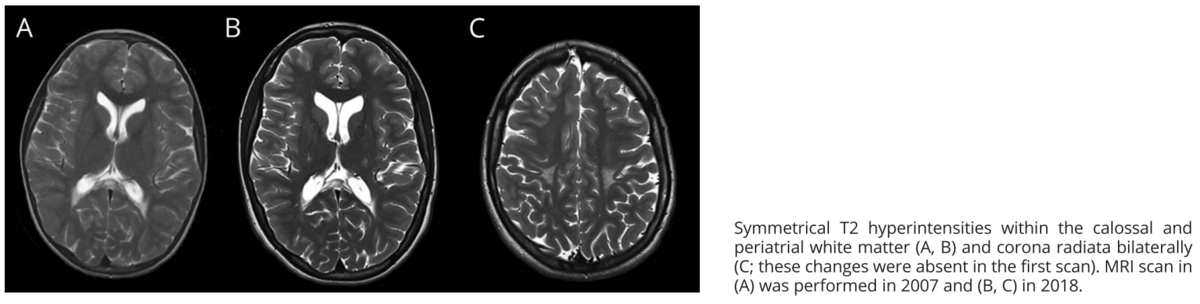
- 脳単純MRI:融合性で左右対称性の脳梁と脳室周囲白質の病変を認め,過去のMRIとは大きな変化はなかった.
MRI所見からは白質脳症が示唆されるが,進行がないことは非典型的な所見であった.

電気生理学的検査
- 神経伝導検査:運動感覚神経とも遠位潜時延長と伝導速度低下があるが振幅は保たれ,過去の検査結果と同様に脱髄性ニューロパチーの所見であった.
- 針筋電図:前脛骨筋で軽度の慢性脱神経変化を認めた.
- 電気生理学的検査では,後天性脱髄性ニューロパチーより,遺伝性脱髄性ニューロパチーが示唆される所見であった.
CMTの遺伝子パネル
PMP22, Cx32, MPZ, ERG2, NFL, GDAP1, LITAF, MFN2, SH3TC2, FIG4は正常.
Question
- 確定診断のため,次に何を検査するか?
Section 3
本症例の鑑別疾患は多様であるため(複合型HSP,白質ジストロフィー),全エクソン解析(WES)が施行された.WESでガラクトセレブロシド(GLAC)に2つの病的な変異を認めた.(母方からdeletion of exons 11-17 (39end) ,父方からc. 857G>A, p.G286D)
血漿GLAC酵素活性は低下.
KDと確定した.遺伝子コンサルトを行い,専門クリニックへ紹介となった.
最終診断
later-onset Krabbe病
Krabbe病
ライソゾーム病.常染色体劣性遺伝.GLAC遺伝子変異.
アメリカでは250,000出生に1人の確立で生じる.
臨床的に2つの表現型に分けられる.
- infantile-onset (<12ヶ月, 85~90%):進行性の神経障害で,2歳までに死亡する.
- later onset (>12ヶ月, 10~15%) :緩徐進行な経過.
さらにlate infantile (~3歳),juvenile (3–8歳),adulthood(8歳~)に分けられる.(本例はjuvenile-onset KD)
infantile-onset KD症例の多くで神経伝導検査で脱髄性ニューロパチーを示唆する所見を認める.